Tratamiento Ortodoncico interceptivo en una paciente con alteración en su Cromosoma X (Síndrome de Turner)
OCTUBRE 2002- 2005. PROGRAMA COVFASE
Para efectos de referencia bibliográfica ha de ser citado de la siguiente manera: Ceglia A., Ruan V., Ulloa R.
"TRATAMIENTO ORTODONCICO INTERCEPTIVO EN UNA PACIENTE CON ALTERACION EN SU CROMOSOMA X (SINDROME DE TURNER)." Revista Latinoamericana de Ortodoncia y Odontopediatria "Ortodoncia.ws edición electrónica marzo 2006. Obtenible en: www.ortodoncia.ws
Ángela Ceglia; Valeria Ruan; Raúl Ulloa
RESUMEN
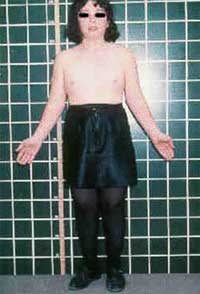
El SINDROME DE TURNER, desde el punto de vista genético es la pérdida total o parcial de un cromosoma X, diagnóstico que solo se puede obtener a través de un cariotipo. Este síndrome se presenta solo en pacientes de sexo femenino, con un gran número de características atípicas tanto fisiológicas como craneobucodentofaciales, psicológicas y sociales, siendo estos pacientes generalmente de talla baja, con problemas de crecimiento y desarrollo y un coeficiente intelectual normal. Se presentan características de cráneo y cara tales como: fusión temprana de las suturas, orejas prominentes en posición baja. Entre las características bucofaciales se presentan mordida cruzada posterior bilateral, mordida abierta anterior, protrusión lingual, paladar estrecho y profundo y deglución atípica. La prevalencia del síndrome de Turner es de 1 por cada 2.500-7.000 niñas nacidas (Hans y Col, 1977) la cifra de la población femenina en Venezuela en 1998 fue de 11.543.186 (OCEI, Censo Junio de 1998),se puede deducir que existían en el país para esa fecha entre 1.649 y 4.617 personas con ese síndrome.
ABSTRACT
Turner Syndrome: Is a genetic disorder resulting in absence of all or part of one sex chromosome X. You can do diagnostic by karyotype in a blood sample, and you can find it only in female patients. Syndrome characterized by specific dysmorphic features, (short stature), skeletal abnormalities, infertility and organ malformation (gonadal dysgenesis): failure to mature sexually, normal Intelligent Quotient. Skull and face may have prominent upturned lobules in ears, presence of a webbed neck. Mouth: palate-high, cleft, gothic; small mandible, complications: crowding of teeth, malocclusion (crossbite, distal molar occlusion, maxillary overset). The exact cause of the disease is not known but it is thought to be a random occurrence affecting approximately one out every 2500 to 7000 live female births (Hans et. all, 1977).
In Venezuela by 1998 census, the number of female with Turner Syndrome was about 1649 and 4617 (Ocei, Censo Junio 1998).
RESUMO
O SINDROME DE TURNER, do ponto genético da vista é as perdas totais ou parcial de a cromosoma X, o diagnóstico que escolhe pode ser obtido com o cariotipo. Este síndrome parece único nos pacientes do sexo feminino, com um grande número de características atípicas tanto fisiológicas como cráneo-buco-dento-faciales, psicológicas y sociais, sendo estes pacientes geralmente da estatura baixo, com problemas do crescimento e o desenvolvimento e um intelectual normal coeficiente. As características do esqueleto e da cara aparecem como: fusão adiantada das suturas, as orelhas proeminentes no protetor abaixam. Entre as características dos bucofaciales parecem desperdiçados cruzado bilateral mais atrasado, mordida aberta precedente, protrusión lingual, degluÇao anormal, paladar entrecho y profundo. Eu síndrome de Prevalencia de Turner es de 1 para cada 2.500-7.000 de as niñas nascida (coluna de Hans y, 1977) cifra feminina del 1998 em Venezuela de poblaÇao de11.543.186 (OCEI, Censo de Junio de 1998), se pede deduzir que estiam em el país para Isa fecha entre 1.649 y 4.617 persoas com ise síndrome.
ANTECEDENTES
En el año 1902, Funke citado por Wellzeguer, H y Col.1, reportó el primer caso comprobable de Síndrome de Turner. El mismo presentaba muchos de los criterios de dicho síndrome, como lo son: pies hinchados y baja estatura en la infancia, amenorrea a la edad de 16 años, carencia de caracteres sexuales secundarios, pterigium colli, implantación baja del cabello en la nuca, orejas grandes con ubicación baja, y paladar profundo y estrecho.
Algunos anatomopatólogos como Olivet en 1923 y Ronderath y Meyer en 1925 citados por Moreno.2, encontraron en algunas pacientes las cintillas ováricas.
Ullrich en 1930, citado por Wellzeguer,.1, observó varios casos los cuales reportó posteriormente y pudo describir a una paciente con marcado linfedema en el primer año de vida, quien posteriormente presentó, baja estatura, pterigium colli y rasgos faciales atípicos, tales como: ptosis de los párpados, orejas prominentes, paladar profundo y arqueado, implantación baja del cabello en la nuca, pecho en escudo y uñas en forma de cuchara. El hipogonadismo no fue mencionado en la descripción original, pero en artículos posteriores escritos por Ullrich en 1949, identificó su caso como Síndrome de Turner.
Sin embargo es mérito de Henry Turner quien en 1938, sistematizó por primera vez la enfermedad que lleva su nombre. Describió 7 pacientes de sexo femenino quienes cursaban con inmadurez sexual, pterigium colli (que lo diferenció del cuello corto del klippel-Feil), baja estatura y cúbito valgo.2
Así mismo, Sharpey y Shafer en 1945, citados por Wellzeguer,1, describieron algunos casos de este síndrome, encontrando gónadas alteradas en uno de ellos, y establecieron el nombre de Síndrome de Turner. En 1947 Jost, así como también Raynaud y Frilley, experimentando con conejos y ratones respectivamente, mostraron que la diferenciación del útero, vagina y genitalidad femenina externa no ocurre si son removidas las gónadas, femeninas o masculinas, o son destruidos durante la vida embrionaria temprana. El desarrollo del ducto de Wolffian no se da si las gónadas masculinas son removidas.
Por otra parte, Castillo en 1947 y Reforzo en 1949, citados por Moreno, 2, publicaron casos de esta entidad, a la que denominaron "síndrome de los ovarios rudimentarios".
En 1954 Polani y Col., citado por Moreno,.2 mediante la determinación del sexo nuclear confirmaron la presencia de cromatina sexual negativa, al no existir más que un cromosoma X. En aquel entonces la ausencia de cuerpos de Barr fue considerado idéntico al sexo nuclear masculino.
Dos años después Polani y Col. usando evidencia proveniente del sexo nuclear y la ceguera hereditaria de colores, sugirieron una constitución X0 para el cromosoma sexual.1
En 1959, Ford y Col, citados por Moreno.2, confirmaron con la introducción del cariotipo que el síndrome se caracterizaba por presentar solamente 45 cromosomas, 44 autosómicos y un cromosoma X.
En 1980, Netter citado por Carrasco 3, refirió que las pacientes que padecen este síndrome son mujeres sexualmente infantiles que exhiben una diversidad de anormalidades congénitas tales como: el cuello palmeado (refiriéndose al pterigium colli) y pecho amplio en forma de peto, las niñas alcanzan una estatura final de 1,48 m y resultan anormalmente cortas al nacer.
Por último, Orrego y Bixler en 1990 citados por Medlena 4 señalaron también a la esterilidad como característica de este síndrome.
ETIOPATOGENIA
Como cualquier otro síndrome cromosómico, si los padres presentan un cariotipo normal, el síndrome de Turner no es hereditario y por lo tanto se puede decir, que se presenta como un hecho aleatorio.1
SINDROME DE TURNER
El Síndrome de Turner, también denominado Disgenesia Gonadal o Agenesia gonadal, representa una variable especial de hipogonadismo hipergonatrófico con fenotipo femenino, ocasionado por la perdida total o parcial del segundo cromosoma sexual. Los cariotipos más frecuentes presentes en esta enfermedad son la Monosomía X Monoclonal, varios Mosaicismos (Mixploides) cada uno con un X-monosómico (X0) (célula asexual) y en otros casos se debe a una Anomalía Estructural del segundo cromosoma X. En el Síndrome de Turner causado por una Monosomía X el sexo nuclear es negativo, mientras que puede ser positivo en el Mixoploide X0/XX y en el Síndrome causado por Anomalías Estructurales del segundo cromosoma X. 1
Los rasgos clínicos más comunes suelen ser: fenotipo externo femenino con carencia de características sexuales secundarias; ausencia en el desarrollo de los senos; escaso vello púbico y axilar; amenorrea primaria; infertilidad (con raras excepciones); gónadas debilitadas; y tamaño y forma de útero y vagina prepuberal.
Otras características que se pueden evidenciar en la infancia son: linfedema de manos, pies y algunas veces de piernas, estatura baja, y ciertos rasgos dismórficos. Las personas afectadas suelen presentar Nevus pigmentado en la piel y en pocas ocasiones telangiectasias en la pared del intestino. Las malformaciones cardiovasculares y renales son frecuentes y el retardo mental no se considera un rasgo patognomónico del Síndrome, pero el coeficiente intelectual de ejecución resulta mas bajo que el coeficiente intelectual verbal.1
MANIFESTACIONES FISIOLOGICAS
Los síntomas clínicos de la Monosomía X Monoclonal, también presenta manifestaciones clínicas y otras variantes, por lo cual el síndrome de Turner es una de las pocas anomalías de sexo en el cromosoma, lo cual puede ser diagnosticado clínicamente en el nacimiento o en la infancia .1
En la clínica destacan tres aspectos importantes:
1.- Talla baja
En cuanto la talla al nacer puede ser de un promedio bajo, por debajo del 3er percentil.
El cromosoma X responsable del crecimiento de estos pacientes se ha estudiado y analizado; por este motivo, cuando un cromosoma X esta alterado o falta, origina alteraciones en el crecimiento. El crecimiento en las niñas con síndrome de Turner se caracteriza porque sigue un patrón característico:
Ya durante el desarrollo intrauterino, durante el embarazo, el médico percibe un lento crecimiento de la niña, de forma que, al nacer, su talla es aproximadamente entre dos y tres centímetros menor que la de las demás niñas.5
A medida que van pasando los años, el crecimiento es cada vez mas lento, y cuando llegan a los 12 años la diferencia de talla con respecto a sus amigas es notorio.5
La mayor diferencia se encuentra en el momento de la pubertad, ya que no aparece el estirón de crecimiento propio de esta época de la vida. La mayoría no producen las hormonas necesarias para el inicio de la pubertad y como consecuencia, se hace mas evidente el retraso del crecimiento En la edad puberal no aparece el estirón de crecimiento propio de esta época de la vida, ya que la mayoría de ellas, no producen las hormonas necesarias para el inicio de la pubertad.5
El crecimiento en las pacientes Turner se mantiene hasta los 19 años y la talla media alcanzada al final de su crecimiento es variable.
2.- Infantilismo sexual
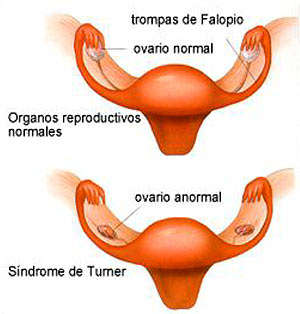
La disfunción más frecuente en un órgano en el síndrome de Turner es la disgenesia ovárica. (ver imagen N 1) Es en el último trimestre del embarazo y en los meses siguientes a la gestación cuando se produce una rápida atresia de los ovocitos y fibrosis del estroma.
Imagen N.1
Disgenesia ovarica http://www.geocities.com/HotSprings/Villa/7158/inform.htm Ovarios
Las gonadotropinas permanecen en niveles altos durante los primeros años de la vida, para luego disminuir a niveles similares a las niñas normales. Al no producirse la pubertad, por fracaso ovárico, aumentan las gonadotropinas hasta niveles de menopáusicas, y por tanto no se produce el desarrollo de los caracteres sexuales secundarios, y las afectas permanecen sexualmente infantiles y con amenorrea primaria.2
Hasta hace poco la infertilidad era definitiva, pero gracias a las investigaciones en la donación de óvulos y su implantación en el útero de la receptora, la maternidad es posible, aunque deben resolverse algunos problemas como cronizar los ciclos hormonales de la donante y de la receptora, así como encontrar el momento optimo para el trasplante de embriones al útero.
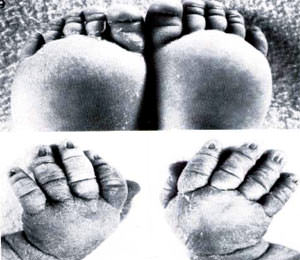
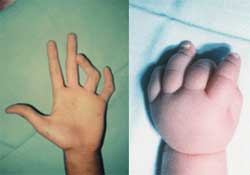
En el momento del parto suele ser frecuente el linfedema de las extremidades (ver imagen N. 2) ó el higroma de la nuca, ambos condicionados por una hipoplasia de los vasos linfáticos superficiales (25% de los casos). Se produce un bloqueo linfático, que en general desaparece durante el primer año de la vida, aunque a lo largo de ella puede dar lugar a fenómenos de edema intermitente y tendencia a la linfangitis.2
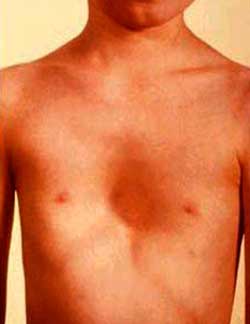
El tórax en coraza y tórax escabatum (ver imagen N. 3), con aumento de la distancia entre las mamilas (tórax en escudo) y no hay desarrollo en los casos típicos. (Ibid)
En niñas jóvenes y adolescentes, también se suele conseguir en sus miembros una característica clásica que es el cubitus valgus (ver imagen N 5) así como el acortamiento del cuarto y quinto metacarpales, pudiéndose conseguir también unas anomalías digitales en los miembros inferiores.3
Imagen N.5 http://www.seepcv.com/atlas/ Tomo2/Creci7/T2C7D10.jpg.
Cubito Valgo.10
En cuanto a la piel y uñas la presencia de nevus es una importante característica a la hora de hacer un diagnostico; pueden ser vistos en la cara, brazos, tórax y en cualquier otra parte del cuerpo. Las uñas son a menudo hipoplásicas y exageradamente convexas y clinodactilia en los dedos. (ver imagen N 6 y 7)
CARACTERÍTICAS CRANEOBUCODENTOMAXILOFACIALES DEL SÍNDROME DE TURNER
Son diversas las características clínicas que las alteraciones cromosómicas determinan en las estructuras craneodentobucofaciales; sin embargo, no siempre se presentan todas en una misma mujer.
Imagen N.8
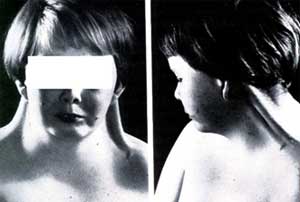
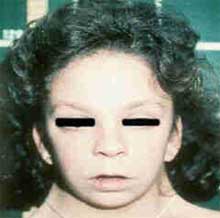
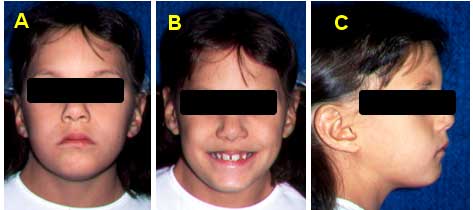
Vallejo, E. Estructuras Craneobucodentofaciales12Los rasgos faciales de las mujeres con síndrome de Turner por lo general son dismórficos; con una cara en forma triangular, comisuras labiales hacia abajo (Ver Imagen 8), hipertelorismo, fisuras palpebrales ubicadas en posición inclinada hacia abajo con epicanto uni o bilateral, ptosis del párpado superior; asimismo, las orejas suelen ser largas, de baja inserción y sobresalientes (Ver Imagen B) y la boca puede tener una ubicación baja. 1 3
En relación a la morfología de los maxilares, se tiene que la dimensión transversal es similar al resto de la población en la mandíbula; pero esta situación no es la misma en el maxilar, ya que en un elevado número de casos se observa una medida disminuida, por esta razón el paladar suele ser estrecho y alto, forma ojival (Ver Imagen N 8), aproximadamente en un 70% de las afectadas. Toda esta situación puede traer como consecuencia maloclusiones, tales como Mordida Cruzada Posterior, la cual puede ser Uni o Bilatral.12
Así mismo, ambos huesos pueden ser más cortos de lo habitual, afectándose más la mandíbula y suelen estar situados en una posición posterior con respecto a su base craneal (retrognatismo), siendo la mandíbula la más afectada, de igual forma la longitud de esta base es más corta; estas anomalías ortodóncicas pueden provocar el desarrollo de disfunción de la articulación temporomandibular.4
Por otra parte, con respecto a los índices de placa, de gingivitis y de caries, que normalmente están relacionados con el grado de higiene bucal, las pacientes con síndrome de Turner suelen presentar bajos niveles los mismos si se les compara con niñas con genotipo normal.12
Posiblemente estos hallazgos podrían deberse a la diferente morfología dentaria en las niñas Turner (determinaría menor acúmulo de placa en las superficies dentarias). Así mismo, según algunos estudios, estas niñas presentan menos problemas de apiñamiento, lo cual podría ser un factor determinante en la poca acumulación de placa bacteriana (Ibíd).
En cuanto a el tamaño dentario, se tiene que en ambas denticiones (temporal y permanente) pueden ser más pequeños de lo habitual, a excepción de los caninos temporales. Existen varias teorías que tratan de explicar el por qué de la disminución en el tamaño de los dientes en las mujeres Turner. En una de ellas se afirma que el menor tamaño es debido a que la capa más superficial de la corona del diente (esmalte) es más delgada de lo habitual y ello se atribuye a la falta del cromosoma sexual. Por otra parte, según algunos investigadores, existe una relación positiva entre el tamaño dentario y la talla; de esta forma, los dientes tienden a ser más pequeños en individuo de baja estatura que en individuos normales o altos (Ibíb).
Con respecto a la erupción dentaria, suele estar adelantada en las niñas que padecen el Síndrome de Turner, es decir, suele ser más precoz de lo habitual. Asimismo, en las relaciones intermaxilares es un hallazgo frecuente el observar una falta de contacto y una separación entre ellos, oclusopatía denominada mordida abierta anterior de igual forma, puede presentarse protrusión incisiva superior (aumento del resalte incisivo) con cierta frecuencia (Ibíd).
De esta forma, el desfase en la relación anteroposterior entre ambos maxilares, además del problema oclusal que ocasiona, produce alteración en la estética del perfil; sin embargo, la mordida cruzada y el paladar ojival pueden ser tratados a través de la expansión palatina y el aumento del resalte incisivo puede corregirse ortodóncicamente. La disminución en los tamaños dentarios, en la mayoría de los casos no es tan intensa como para producir problemas estéticos, por lo que no requiere tratamiento (Ibíd).
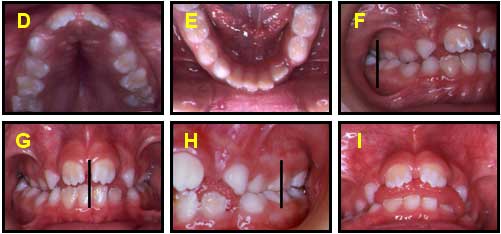
REPORTE DEL CASO FOTOS EXTRAORALES (INICIALES): DE FRENTE (A), SONRIENDO(B),PERFIL (C)FOTOS INTRAORALES (INICIALES): Maxilar superior D, Max. Inferior E, Oclusión lado derecho F, oclusión de frente G, Oclusión lado Izquierdo H, Oberjet de 5 mm I. Diagnóstico
Dental: Clase I
Esqueletal: Clase I
Lista de problemas:
Mordida abierta anterior.
Mordida cruzada posterior bilateral
Paladar profundo y estrecho
Protrusión lingual
Deglución átipica
Respiración bucal
Objetivos del tratamiento:
Expansión maxilar
Descruzamiento de mordida cruzada posterior bilateral
Cierre de mordida abierta
Control de hábitos
Pronóstico:
Reservado
Plan de tratamiento:
Aparatología removible activa modificada. Superior: 2 tornillos expansores, mantenedores de espacios, arco de Hawley, escudos vestibulares, pistas planas. Inferior: Arco de Hawley, pistas planas, mantenedores de espacio.
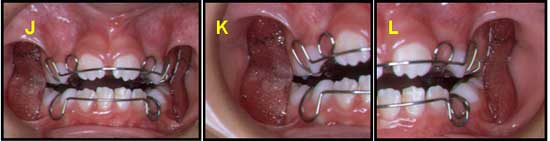
INSTALACION DE APARATOLOGIA: Oclusión de frente con aparatología J, Oclusión lado derecho con aparatología K, Oclusión lado Izquierdo con aparatología L
Fecha de evaluación: PRIMER CONTROL 30 de Abril de 2003
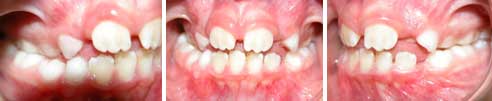
FOTOGRAFÍAS DE PRIMER CONTROL: Oclusión lado derecho con aparatología M, Oclusión de frente con aparatología N, Oclusión lado Izquierdo con aparatología O, CON APARATOS: M1, N1, O1.
M, N, OM1, N1, O1
Fecha de evaluación: SEGUNDO CONTROL 2 de Julio de 2003
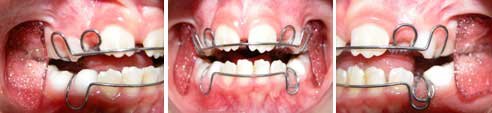
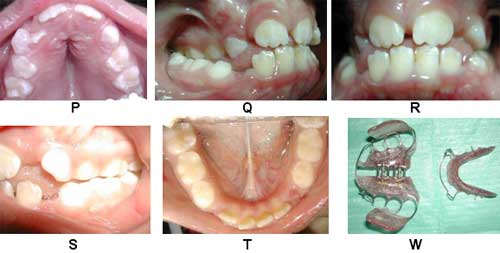
FOTOGRAFIAS SEGUNDO CONTROL: Maxilar superior P, Oclusión lado derecho Q, oclusión de frente R, Oclusión lado Izquierdo S, Max. Inferior T, Segunda Aparatología W.
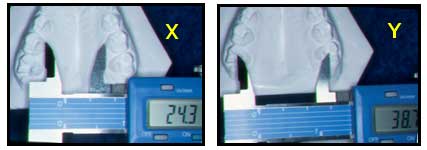
ANALISIS DE BOGUE (Primer Control)
X
ANALISIS DE MAYORAL (PrimerControl)
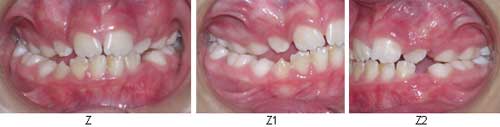
Y FOTOS TERCER CONTROL: Oclusión de frente Z, Oclusión lado derecho Z1, Oclusión lado Izquierdo Z2
Resultados:
Primer control: Al análisis de los modelos, en el índice de Bogue y mayoral se evidencio un aumento de 1mm.
Segundo control: Cierre de la mordida abierta anterior, descruzamiento de la mordida cruzada posterior del lado izquierdo. Al examen de los modelos, se observó un aumento del índice de Bogue de 1 mm e Indice de Mayoral de 2mm.
Tercer Control: Eliminación del hábito de protusión lingual y deglución atípica. Erupción de laterales y caninos superiores. Al examen de modelos , se observó un aumento en el índice de Bogue y Mayoral de 1mm.
Conclusiones:
El tratamiento Interceptivo que se le aplicó a la paciente, dió resultados positivos en un período corto de tiempo. Los cambios mas significativos que se han evidenciado hasta la fecha son la expansión de los maxilares, lo que ha permitido el descruzamiento de la mordida cruzada del lado izquierdo de la paciente, así como el cierre de la mordida abierta anterior, la eliminación del hábito de protusión lingual y deglución atípica. La paciente actualmente continua con el tratamiento.
REFERENCIAS BIBLIOGRAFICAS
WELLENGUER, Hans; SIMPSON, Jane (1977). Chromosomes of Men. Spaspics International Medical Publication. Londres, Inglaterra.
CARRASCO, William. Síndrome de Turner. Clínica al Día. Año 7N0. 2- 1998. Pág. 76-90.
MEDLÉNA, Melida; SZILÁGYI, Zoltán; KESZTHELYI, Gusztáv (1998). Síndrome de turner: Revisión de la literatura y reporte de un Caso. Journal of Dentristry. Edición en Español. Pág. 62-64.
GROHMANN, Ulrike. 2002. Aparatología en Ortopedia Funcional. Atlas Gráfico. Editorial AMOLCA., Caracas, Venezuela. Primera Edición. http://www.odontocat.com/tratortofuncional.htm. Especialidades de Ortodoncia. Aparatología Funcional. Fecha de Consulta: 25/04/2003.