
Introducción: La hendidura orofacial corresponde a una de las malformaciones orofaciales más comunes en los defectos de nacimiento y su desarrollo es previo a la novena semana de gestación y su etiología es multifactorial. El trabajo en equipo y su multidisciplinariedad es esencial para evitar las secuelas prevenibles. Metodología: Búsqueda bibliográfica en PUBMED y Google Scholar en el año 2020 con los criterios:” (orofacial cleft) AND (pathogenesis) OR (etiology)”. Se incluyeron y revisaron 14 artículos. Resultados: Existen una serie de factores genéticos (genes principales, genes menores) y ambientales responsables de la patogénesis de las hendiduras orofaciales y se ha descrito que se pueden manifestar como un defecto de cierre sindrómico o defecto de cierre no sindrómico. Hoy en día, la teoría más aceptada corresponde a un terreno genético predispuesto en el cual actúan agentes externos de manera que la variación genética modula el riesgo de los defectos. Conclusión y discusión: Las Hendiduras orofaciales son de etiología multifactorial involucrando factores genéticos y ambientales, y se presentan como un defecto no sindrómico en un 70% de los casos. Se debe ampliar el espectro fenotípico de las hendiduras aisladas ya que un individuo que aparentemente no está afectado puede albergar al menos uno de los sub-fenotipos y debería considerarse como un posible portador de genes.
Palabras clave: cleft palate, etiology, embryology, genetics.
Introduction: The orofacial cleft corresponds to one of the most common orofacial malformations in birth defects and its development is prior to the ninth week of gestation and its etiology is multifactorial. Teamwork and its multidisciplinarity is essential to avoid preventable sequelae. Methodology: Bibliographic search in PUBMED and Google Scholar in 2020 with the criteria: "(orofacial cleft) AND (pathogenesis) OR (etiology)". 14 articles were included and reviewed. Results: There are a series of genetic factors (major genes, minor genes) and environmental factors responsible for the pathogenesis of orofacial clefts and it has been described that they can manifest as a syndromic closure defect or non-syndromic closure defect. Today, the most accepted theory corresponds to a predisposed genetic terrain in which external agents act in such a way that genetic variation modulates the risk of defects. Conclusion and discussion: Orofacial clefts are of multifactorial aetiology involving genetic and environmental factors, and present as a non-syndromic defect in 70% of cases. The phenotypic spectrum of isolated clefts should be broadened as an apparently unaffected individual may harbor at least one of the sub-phenotypes and should be considered as a possible gene carrier.
Key words: cleft palate, etiology, embryology, genetics.
Los defectos de cierre orofaciales corresponden a defectos congénitos (Escobar et al. 2013) que se desarrollan previo a la novena semana de gestación y pueden ser clasificados anatómicamente como aquellos que afectan al paladar secundario (paladar blando y duro), denominados paladar hendido aislado; y aquellos que afectan al paladar primario y se acompañan de defectos de cierre del labio, acompañados o no de paladar hendido. Esta distinción es importante porque se apoya sobre bases embriológicas 1.
Esta alteración es de etiología multifactorial, e involucra factores genéticos y ambientales. La mayoría de este tipo de patologías se presenta como un defecto no sindrómico en un 70% de los casos, y el resto de hendiduras se asocian con alteraciones adicionales en los defectos de tipo sindrómico 2.
En la actualidad, y considerando que la biología molecular está cada día más al alcance de todos, es necesario que como profesionales de la salud tengamos conocimiento de las bases moleculares y genéticas de estas alteraciones para así poder orientar a pacientes adultos que presenten algún fenotipo subclínico, e informarles a tiempo las posibilidades de que su descendencia pudiera presentar algún tipo de hendidura 3. Esto es muy relevante ya que el diagnóstico precoz reduce las angustias de los padres y del personal de salud al momento del nacimiento. Permite, además, tomar las medidas necesarias para garantizar las mejores condiciones al recién nacido con esta malformación. Luego del diagnóstico pre-natal se puede realizar un abordaje integral del grupo familiar durante el embarazo, lo cual ayudará a conocer las dimensiones exactas del problema, informarse, planificar y anticipar el tratamiento 4.
El trabajo en equipo y su multidisciplinariedad es esencial para el éxito del tratamiento y poder evitar las secuelas prevenibles. En el equipo participan cirujanos, enfermeras, fonoaudiólogos, genetistas, kinesiólogos, odontólogos, ortodoncistas, otorrinolaringólogos y psicólogos. El éxito de los resultados dependerá de la experiencia del equipo multiprofesional, planificación, investigación y seguimiento metódico de los casos, auditoría de los tratamientos y capacitación constante, independiente de la labor asistencial 5. Es por ello que es de suma importancia que los(as) cirujano dentistas y en especial los(as) ortodoncistas, odontopediatras y cirujanos(as) maxilofaciales, tengan una buena base con respecto a la patogenia de las hendiduras orofaciales (HOF).
El objetivo de esta revisión es ordenar y sintetizar la bibliografía disponible en la actualidad sobre etiopatogénesis de las hendiduras orofaciales con el fin de facilitar el entendimiento y acceso a esta información por parte de los cirujanos dentistas, debido a la relevancia de su participación en el equipo multidisciplinario.
En el año 2020 se realiza una búsqueda bibliográfica según los parámetros indicados en la Figura 1. En una primera instancia los artículos fueron seleccionaron por atingencia del título, luego por lectura del abstract y finalmente con la lectura del artículo completo. El criterio de selección fue estudios que describieron alguno de los factores etiopatogénicos de las fisuras orofaciales sindrómicas o no sindrómicas, ya sea genéticos o ambientales, así como su relación con las distintas etapas de la embriogénesis.
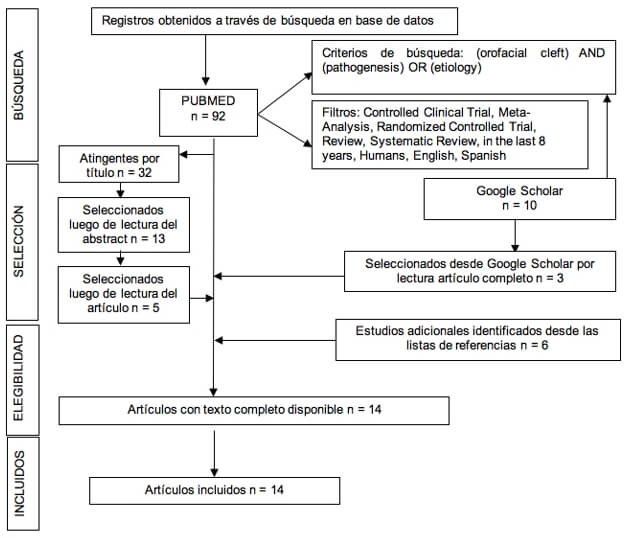
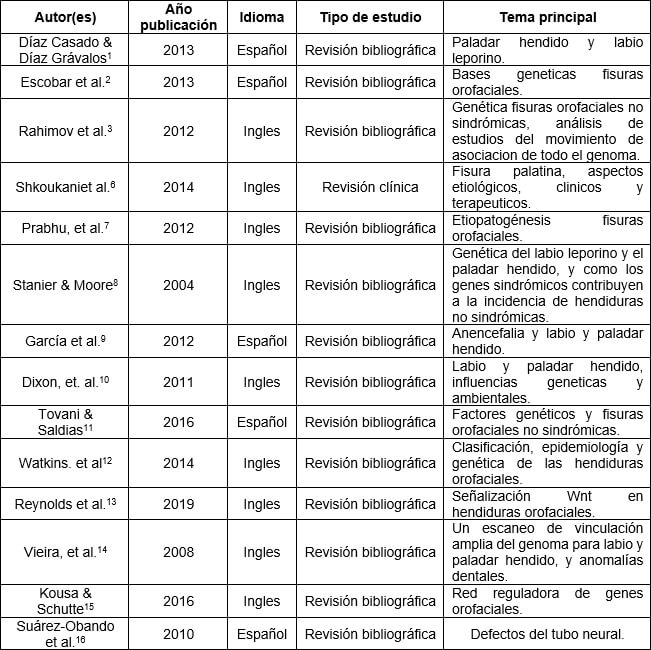
Se obtuvieron 14 estudios que cumplían con los criterios de inclusión (Tabla I). Luego de su lectura completa se seleccionó, sintetizó y agrupó la información en los siguientes puntos para su mejor comprensión:
El desarrollo de la cara comienza con la migración de las células de la cresta neural durante la 4ª semana de gestación para formar los procesos frontonasal, maxilares y mandibulares 3,6.
Durante la 5ª semana el proceso frontonasal dará origen a los procesos nasales mediales y laterales, los procesos nasales mediales se fusionarán durante las siguientes dos semanas y formarán el paladar primario 3.
Al final de la 6ª semana, se fusionan los procesos nasales mediales entre sí formando el filtrum labial y luego estos se fusionan con los procesos maxilares formando el labio superior y el paladar primario 6.
Durante esta fusión los procesos se contactan, se adhieren y se produce una lámina epitelial media que desaparece rápidamente por apoptosis, permitiendo la confluencia de las células mesenquimales y la finalización de la fusión 2,6.
Justo antes de la fusión, el proceso nasal tiene un peak de crecimiento por división celular, lo que aumenta su susceptibilidad a daños por teratógenos que resultarían en fallo de la fusión. Fracasos en la fusión en esta etapa causaría las hendiduras labio palatinas (HLP)6.
Los procesos palatinos crecerán verticalmente desde los procesos maxilares durante la 6ª semana por proliferación celular 2,3,6 y se elevarán a su posición horizontal por encima de la lengua durante la 7ª semana de gestación, donde seguirán creciendo hasta entrar en contacto durante la 8ª semana para finalmente fusionarse en la línea media utilizando mecanismos de migración y apoptosis celular 3.
Hendiduras del paladar secundario se deben a fracasos en la elevación de estos procesos palatinos o a fracasos en el contacto, adhesión y fusión entre ellos mismos. Lo normal es que las células de la línea media se fusionen rápidamente al contactarse luego de un proceso de apoptosis y diferenciación celular 2,6.
A la 10ª semana se puede observar fusión completa del paladar secundario, labio superior y septo nasal, terminando completamente la formación a la 12ª semana 6.
El desarrollo orofacial es una secuencia de eventos estrictamente coordinada espacio temporalmente que involucran migración, proliferación, diferenciación, fusión y apoptosis celular 3. Cualquier alteración en alguno de estos mecanismos puede resultar en una HOF 2,3.
La expresión de las HOF es heterogénea y la variación en el riesgo puede estar determinada por una serie de factores como: genes principales, genes menores y factores ambientales 7.
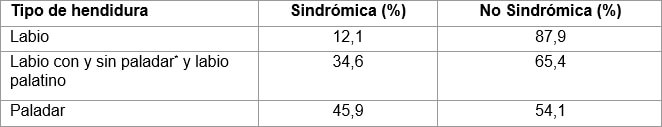
Clínicamente, cuando el defecto de cierre orofacial aparece con otras malformaciones dentro de patrones reconocibles, se clasifica como defecto de cierre sindrómico. Si aparece como un hecho aislado o si no es posible identificar el síndrome, se denomina defecto de cierre no sindrómico (Tabla II). El número de síndromes que presentan defectos de cierre es muy elevado, actualmente se mencionan entre 300 y 500 síndromes 1,2.
La teoría más aceptada es que los agentes externos actúan sobre un terreno genético predispuesto, interactuando con la genética materna y fetal, de tal forma que la variación genética es la que modula el riesgo de defectos 1.
Las hipótesis genéticas para la etiología de HOF han evolucionado considerablemente, pero a pesar de existir numerosas y extensas investigaciones, ningún patrón mendeliano simple de herencia ha sido fácilmente identificado. Esto ha llevado a la propuesta de diferentes autores de una variedad de modos genéticos de herencia que incluyen: dominancia, recesividad, vinculación sexual y diversas condiciones de modificación, como penetrancia incompleta y expresividades variables 7.
Se han asociado una variedad de moléculas de señalización en la formación de los primordios faciales, la diferenciación epitelial y la remodelación de las estructuras (Figura 2). Estas incluyen, entre otras, moléculas de la matriz extracelular y factores de crecimiento, que actúan como señales inductivas como Sonic hedgehog (Shh) en el epitelio del borde medial, proteínas morfogenéticas óseas (Bmp), factores de crecimiento de fibroblastos (Fgf) y miembros de la súper-familia del factor de crecimiento transformante β (Tgfβ). El gen homeobox Msx1, decodificador de factores de transcripción e involucrado en la etiología de las agenesias dentarias, se expresa en los primordios faciales, y se requiere para la expresión de Bmp2 y Bmp4 en la mesénquima palatina y Shh en el epitelio del borde medial 8.
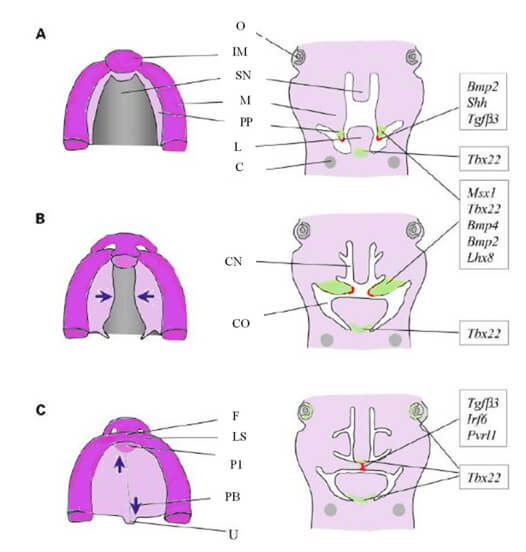
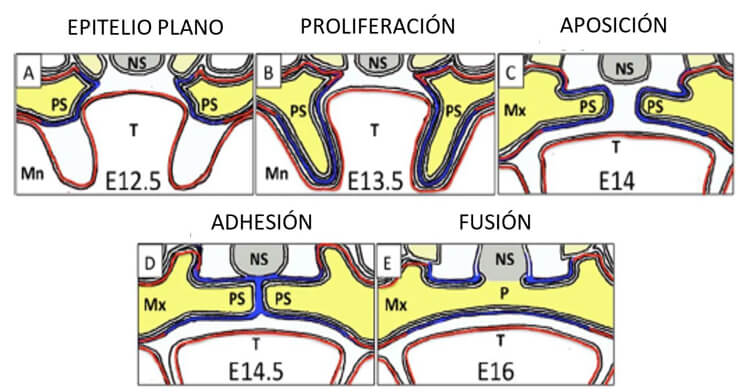
A: Secciones horizontales y coronales emparejadas de cabezas de ratón en E12.5 (semana humana ≈7). Las prominencias nasales medias se fusionan para formar el segmento intermaxilar (IM) mientras que las partes laterales del labio superior se forman a partir de las prominencias maxilares (PM). Los procesos palatinos (PP) también brotan de las prominencias maxilares (PM) y crecen verticales hacia la lengua (L).
B: En el ratón E13.5 (semana humana ≈8), los procesos palatinos sufren remodelación epitelial y se elevan a una posición horizontal por encima de la lengua. Las flechas moradas indican la posición del contacto inicial de los procesos y la fusión.
C: Con el ratón E14.5 (semana humana 9-10), el segmento intermaxilar se convierte en el filtrum (F) del labio superior (LS) y el paladar primario (P1). Los PP se han fusionado en ambas direcciones, anterior y posterior (flechas moradas), junto con el tabique nasal. La unión epitelial se rompe y la osificación comienza en el paladar anterior (duro). Los sitios de expresión para algunos de los genes clave que afectan la palatogénesis se indican en las secciones coronales: el sombreado verde corresponde a la expresión de Tbx22; el rojo indica el epitelio del borde medial palatino caracterizado por la expresión de Tgfb3. Los genes como Pvrl1 y p63 se expresan a través del epitelio oral. SN=Septum Nasal; O=Ojo; CM = cartílago de Meckel; CN = cavidad nasal; CO = cavidad oral; PB = paladar blando; U=úvula. (Adaptado de Stanier & Moore 8)
La aberración en la programación y secuencia de alguno de estos genes es probablemente el origen de la patogénesis de las HOF 9.
Otros genes importantes son el FOXE1, involucrado directamente con el desarrollo embrionario y el PVRL1, decodificador de moléculas de adhesión celular 10–12.
Las HOF sindrómicas y no sindrómicas se han atribuido a mutaciones de varios genes del componente de señalización Wnt, esto por su rol de la morfogénesis craneofacial, incluyendo las fisuras orofaciales 9,13.
Finalmente, los microARNs tienen un papel muy importante en la regulación del desarrollo embriológico debido a sus perfiles de expresión, los cuales son muy dinámicos durante el desarrollo embrionario, donde cualquier alteración en su expresión puede perturbar la embriogénesis, organogénesis, y la homeostasis del tejido, junto con el ciclo celular; teniendo un papel clave en la regulación de la diferenciación neural de células madre 9,13.
Aproximadamente el 70% de todos los casos de HOF y el 50% de los casos de hendidura palatina (HP) se consideran no sindrómicos 10.
La HP aislada se podía heredar como un rasgo dominante con una penetrancia muy baja, mientras que la HLP posiblemente se transmite por un gen de penetrancia variable que podría actuar como un gen recesivo o dominante, dependiendo de los antecedentes genéticos del individuo 3,7.
Algunos datos epidemiológicos sugieren que la hendidura labial, puede tener características etiológicas únicas, incluidas fuertes asociaciones genéticas, mientras que algunos individuos sólo con HP, muestran evidencia de hendidura labial subclínica. Sin embargo, esta amplia subdivisión de defectos anatómicos es consistente con los distintos orígenes de desarrollo del labio / paladar primario versus el paladar secundario 10.
Sin embargo, los estudios de vinculación para trastornos complejos, como lo son estas hendiduras, han tenido un éxito limitado debido a la alta heterogeneidad genética y fenotípica observada 3.
De todas maneras, se cree que algunas características subclínicas sutiles pueden ser parte de un fenotipo de hendidura aislado "extendido". Por ejemplo, los defectos ocultos en el músculo orbicular del labio superior (también llamada hendidura labial sub-epitelial) pueden representar la forma más leve de una hendidura aislada (Figura 3). De hecho, los familiares no afectados de individuos con HP abierta tuvieron el doble de probabilidades de tener defectos en el músculo orbicular que aquellos sin antecedentes familiares 3,10.
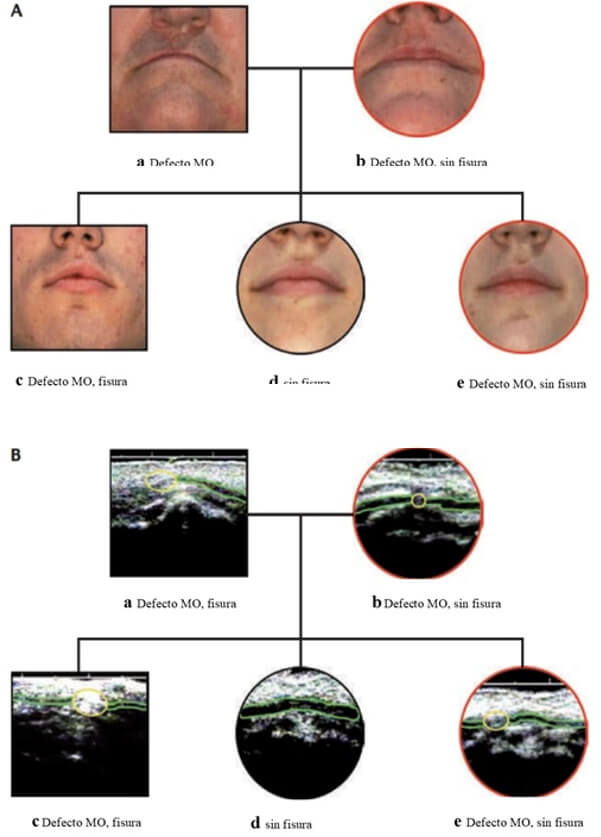
A: Fotografías de la región del labio superior para cada miembro de una familia nuclear con dos miembros de la familia afectados con HL/P no sindrómica. Los otros miembros no tienen defectos visibles desde el exterior, pero dos de ellos tienen defectos subclínicos del músculo orbicular (MO) (símbolos de pedigrí encerrados en rojo).
B: Los ultrasonidos del labio superior de cada miembro de la familia. Observe las interrupciones en el MO en las dos personas con HL/P y de las dos personas sin manifestación externa (símbolos de pedigrí encerrados en rojo)
(Adaptado de Dixon et al.10).Si además de los defectos musculares, se considera el papel de las anomalías dentales, como agenesias, las anormalidades estructurales del cerebro, la morfología craneofacial, como craneosinostosis, y los patrones en espiral en el labio inferior, como indicadores de un riesgo genético subyacente, es muy prometedor en el análisis más fino de las relaciones entre estos sub-fenotipos 3,14.
Ampliar el espectro fenotípico de las hendiduras aisladas para incluir estos sub-fenotipos (Figura 4) puede ayudar a explicar los problemas de penetrancia incompletos que se han observado en varias familias en las que se transmite una mutación aparentemente etiológica de un padre sin HP evidente 3.
Vieira et al. informan, por primera vez, una serie de búsquedas en todo el genoma de loci de susceptibilidad a la hendidura utilizando anomalías dentales como sub-fenotipo de hendiduras. Este enfoque puede ayudar en la identificación de variantes genéticas, lo que sería un paso crucial que podría permitir mejores estimaciones de los riesgos de recurrencia para familias individuales 14.
El gen más citado y conocido entre todos los candidatos para la ocurrencia de estas malformaciones en la actualidad, es el factor regulador de interferón del gen 6 (IRF6) 3,10,12, esto porque presenta mayor relación con la ocurrencia de hendiduras y por estar involucrado en cerca de 12% de los casos de HOF no sindrómicas 6,10. Investigaciones han demostrado que los ratones mutantes IRF6 exhiben una epidermis hiperproliferativa que no puede experimentar diferenciación terminal, lo que conduce a múltiples adherencias epiteliales que pueden ocluir la cavidad oral y provocar HP 10,15 (Figura 5).
Las causas de los síndromes de malformación humana generalmente se dividen en 3 categorías: (1) Anomalías cromosómicas y reordenamientos genómicos; (2) Trastornos mendelianos o de un solo gen; e (3) Interacciones complejas de factores genéticos, ambientales y estocásticos, referidos como multifactoriales. Debido a que los teratógenos interrumpen la morfogénesis embrionaria y fetal normal, y pueden interactuar con factores genéticos predisponentes, los factores ambientales siempre deben considerarse en una discusión sobre las causas de los síndromes de HOF. Actualmente se han descrito alrededor de 500 síndromes que dentro de sus manifestaciones pueden presentar HOF y su variabilidad de expresión junto con la penetrancia incompleta son características de estos fenotipos10,12.
De ellos, se estima que un 50% son autosómico recesivo, 40% de ellos son autosómico dominante y el 10% restante está ligado al cromosoma X (Prabhu et al.; Shkoukani et al.).
El labio y el paladar hendidos son malformaciones comunes en ciertas alteraciones de origen genético, como es la trisomía deI par 13 o síndrome de Patau. Los niños afectados presentan labio y paladar hendidos, polidactilia, defectos oculares y sordera, generalmente mueren al poco tiempo de nacer17.
El gen TBX22 se ha relacionado en la forma heredada mendeliana de HP ligada al X (CPX), que exhiben un alto grado de penetrancia. Además de la HP o, en algunos casos, la úvula bífida o ausente, la mayoría de estos pacientes también muestran anquiloglosia 8 (Ilustración 1).
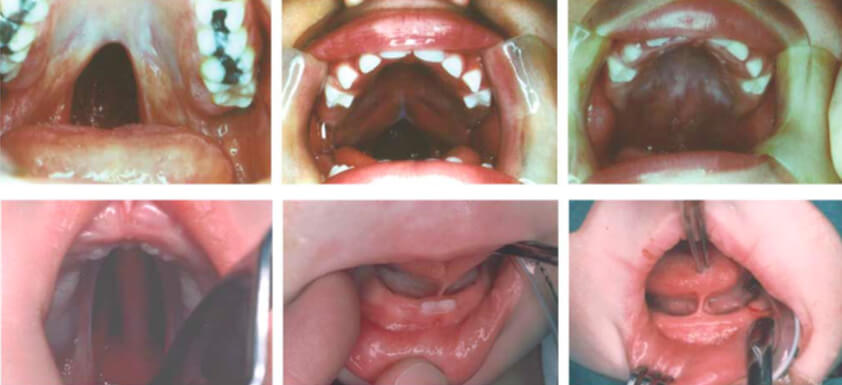
Panel superior: la imagen de la izquierda muestra el paladar blando hendido aislado en la madre con una deleción de base única en la región de codificación de TBX22. El hijo (centro) tiene una hendidura parcial menos severa pero también tiene anquiloglosia (no se muestra). La hija (derecha) tiene una hendidura parcial similar a la de su hermano, pero se muestra en el proceso de reparación. Al igual que la madre, no tiene anquiloglosia y es una fenocopia para la HP no sindrómica.
Panel inferior: el fenotipo CPX masculino clásico es una hendidura completa del paladar secundario (imagen izquierda) y anquiloglosia (imagen central). La imagen de la derecha muestra la anquiloglosia en un hombre relacionado con la úvula bífida menos grave (no se muestra). Su hermano también porta la misma mutación pero tiene anquiloglosia solo sin HP (no se muestra) (Tomada de Kousa & Schutte 15).
Las mutaciones en IRF6 causan dos trastornos de hendidura de herencia autosómica dominante conocidos como Síndrome de Van der Woude (Tabla III) y el Síndrome de Pterigion Poplíteo3,6.

La secuencia de Robin (comúnmente conocida como Síndrome de Pierre Robin [PRS]) merece una mención especial ya que los conceptos actuales la clasifican como un complejo de malformaciones o una secuencia, en reconocimiento de la secuencia embriológica de los eventos involucrados. La PRS generalmente se define por la tríada de micrognatia / retrognatia, HP (generalmente en forma de U pero puede tener forma de V) y glosoptosis (posición posterior anormal de la lengua)12.
Se describe que muchos de los factores medioambientales están aún en estudio, principalmente asociado a su mecanismo de acción. Algunos de ellos inhiben la cadena de transporte de electrones y los efectos de hipoxia en el desarrollo de la prominencia facial en ratones y con ello mostró que estos eran potentes inductores de HP y labio hendido en mamíferos 7.
Se describe que, a mayor edad de la madre, mayor son las probabilidades de incidencia de HOF ya que hay mayor probabilidad de la formación de un cigoto defectuoso. También se menciona que la edad avanzada del padre incrementa el riesgo de paladar hendido aislado 1,7.
Existe una asociación entre alcoholismo y el embarazo con un patrón de anormalidades conocido como Síndrome de Alcoholismo fetal. El consumo de alcohol durante el embarazo puede causar efectos teratogénicos como retraso en el crecimiento, deficiencia mental, defectos cardiovasculares, además de que ocasionalmente se asocia a las HOF 1,7.
Por otro lado, se indica la inconsistencia en el consumo de alcohol durante la gestación como factor de riesgo de las HOF, sin embargo, algunos sugieren que el consumo de alcohol por “atracones” (altas dosis de alcohol en periodos cortos de tiempo) aumentan el riesgo, y esto está respaldado por asociaciones con la variación en el gen de la deshidrogenasa alcohólica (ADH1C) 10.
Se estima que la asociación entre el consumo de cigarro durante el embarazo y el riesgo de tener un hijo con HOF no sindrómica, en el cual sugirió que el consumo de cigarro durante el primer trimestre de gestación se asocia con un mayor riesgo de tener un hijo con HOF (11% de HLP y 12% de HP). Se piensa que el consumo de cigarro podría influir en el desarrollo embrionario produciendo hipoxia del tejido, lo que impide el crecimiento tisular, principalmente en el desarrollo palatino 7. Por otro lado, se describe que la asociación entre el consumo de cigarro y la HOF podría ser mayor a un 20% 1.
Dixon et al, en su revisión señalan que la exposición de la madre al tabaco durante el periodo previo a la concepción, aumenta el riesgo ya que aumenta la posibilidad de que los genes en ciertas vías metabólicas podrían tener un rol en el desarrollo de la HLP. Ellos describen que los marcadores en los genes glutatión S-transferasa-01 (GSTT1) u óxido nítrico sintasa 3 (NOS3) parecen influir en el riesgo de HLP en presencia de madres fumadoras. Estos hallazgos proporcionan evidencia de la importancia de la interacción genético-ambientales en las HLP 10.
Se ha descrito que algunas drogas pueden estar asociadas con un mayor riesgo de tener un hijo con HOF, dentro de las cuales se encuentran agentes quimioterapéuticos para el cáncer (Aminopterina, metotrexato, ciclofosfamida, procarbazina y derivados del ácido hidroxámico), anticonvulsivantes (fenitoína, trimetadiona, parametadiona, carbamazepina, ácido valproico, mysoline y fenobarbital), antagonistas de ácido fólico, deficiencia de ácido fólico, deficiencia de vitamina A y B, radiación, retinoides, antieméticos, hidrocortisona, opiodes, salicilatos (aspirina), diazepam, ácido bórico y antibióticos (nitrofurantoína) 1,7. A continuación, se nombran algunas de ellas:
Recientemente un estudio constató un incremento del riesgo de defectos de cierre en hijos de mujeres con IMC mayor a 30mg/kg2 1,10. También se menciona que la Diabetes Gestacional podría aumentar el riesgo de defectos en el tubo neural 16.
La presencia de contaminantes en el aire, incineradoras de basura en las cercanías o productos de desinfección en el agua (básicamente cloro) presentaron una asociación baja con respecto a la aparición de HOFl 1.
Se ha considerado un factor de riesgo en la aparición de defectos en el cierre orofacial ya que las infecciones virales activan interferones, y se menciona que existe una asociación entre la presencia de defectos de cierre con el gen IRF6 1,10.
También se describe que la anorexia, el estrés, la hipertermia y matrimonios consanguíneos podrían aumentar el riesgo de HOF 1,10. Sin embargo, todavía no existe consenso sobre los efectos nocivos de estos factores por lo que Dixon propone que se requiere de estudios de cohorte prospectivos lo suficientemente grandes como para medir los efectos sobre esta alteración 10.
El periodo previo a la 9ª semana de gestación se considera como el más sensible a sufrir fallas de unión, especialmente entre los procesos palatinos que se cierran completamente en la 12ª semana, lo que se podría manifestar como una HOF.
Existe una correlación entre el desarrollo craneofacial y el cierre del tubo neural en las primeras semanas de gestación, por ello mientras antes ocurren las alteraciones, es más probable que se manifiesten otras malformaciones a parte de las HOF, lo que se denomina síndrome.
Las HOF son malformaciones de etiología multifactorial que involucran factores genéticos y ambientales, y se presentan como un defecto no sindrómico en un 70% de los casos.
La evidencia respalda que los factores genéticos están asociados con la HOF y su variación en riesgo se determina por factores como: genes principales, genes menores y factores ambientales.
En las hendiduras de origen no sindrómico, el gen más citado es el IRF6, y el MSX1, que además está involucrado en la etiología de las agenesias dentarias.
Las mutaciones en IRF6 son las más comunes y causan dos trastornos de hendidura con herencia autosómica dominante conocidos como Síndrome de Van der Woude y el Síndrome de Pterigion Poplíteo. También es importante el gen TBX22 debido a su alta penetrancia relacionada en la forma heredada mendeliana de HLP ligada al cromosoma X y asociada en algunos casos a anquiloglosia.
Respecto a las hendiduras de origen sindrómico, se han descrito más de 500 síndromes y se menciona que un 50% son de herencia autosómica recesiva, 40% autosómica dominante y el 10% restante estaría ligado al cromosoma X.
Es importante ampliar el espectro fenotípico de las hendiduras aisladas para incluir los sub-fenotipos mencionados, ya que las características subclínicas o sub fenotipos pueden ser parte del espectro general de la hendidura aislada, donde un individuo que aparentemente no está afectado puede albergar al menos uno de los sub-fenotipos y debería considerarse como un posible portador de genes.
La patogénesis de las diferentes formas de HOF se deben seguir estudiando, especialmente los factores medioambientales ya que son más modificables y permiten prevenir la aparición de estas mismas.